

Transcriptional Landscape using RNA Sequencing of Long and Short RNAs and Identification of 5’Terminii with each Isoform: The information content resident in genomes can be observed in the transcriptional products produced. This transcriptional activity is pervasive across most genomes. The RNAs produced are not only numerous but also represents a wealth of varieties of transcripts that vary by length (long >200 nt and short <200nt) as well as complex non-contigous representations of the linear genome. Recently, an appreciation that most of the RNA made by genomes do not code for protein has prompted a searches focusing on if and what functional roles the non-coding RNAs have. While this search has been most intense in animal cells leading to the identification of 15,000 new non-coding RNA genes, recent studies in plant cells are revealing remarkable new functions for non-coding RNAs.
One of the goals of the MaizeCODE project is to contribute to the identification of the functional coding and non-coding RNAs starting with five tissues obtained from four strains of maize. This effort begins with the cataloguing and mapping the sites of transcription occurring in each tissue obtained from each strain (Table 1). RNAs transcribed at these loci will provide information concerning the cis-acting regulatory regions by mapping of transcriptional start sites (TSS) using RAMPAGE, a technique developed to identify the 5’ capped structure and its downstream sequences. In so doing the 5’ termini for each isoform and its promoter region can be identified. Additionally, transcriptional termination sites (TTS), RNA splices sites, the possible product-precursor relationships between mapped long and short RNAs will be determined. Clues to whether a RNA (coding and non-coding) is functional can be gleaned by analysis of the transcript sequences to determine how evolutionarily conserved the sequence are and the similarity of the processed isoforms are among the tissues it is expressed in.
The combination of the genome-wide transcriptional activity and identity of the RNAs expressed along with the regulatory element consisting of transcription factor binding sites, histone and DNA modification sites set in the context of the genome sequences of each of the strains being studied provides the foundation for an encyclopedia of functional elements operating in each of the tissues obtained from each species.

Key personnel: Thomas Gingeras (PI), Alex Dobin (computation), Jorg Drenkow (RNA sequencing), Cassidy Danyko (RNA sequencing)
Phone: 516-422-4109 | Email: gingeras@cshl.edu | Website: gingeraslab.cshl.edu

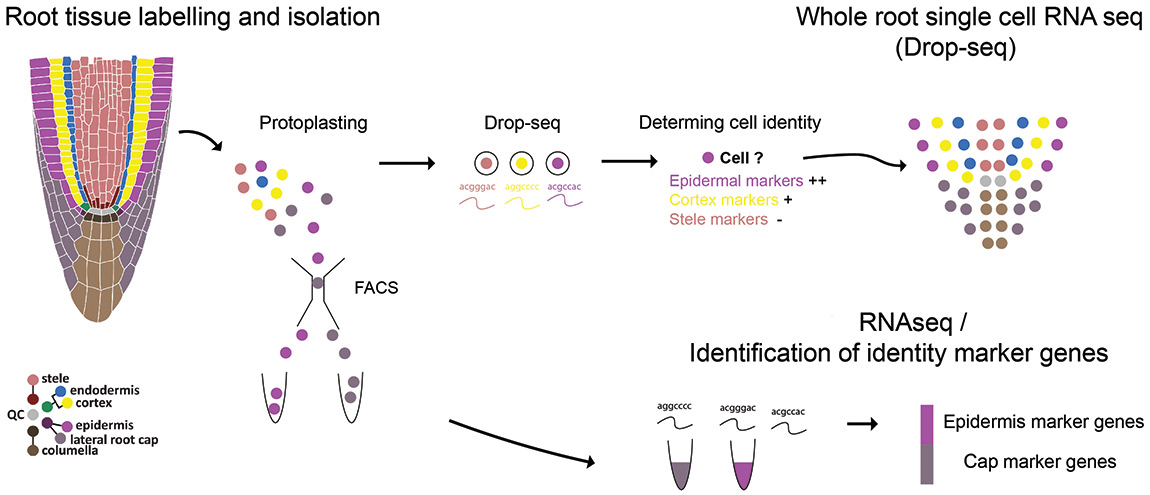
Our group is focused on identifying the genetic regulators that control cell differentiation and specify cell identity during plant organ development. To identify these master regulators, we have isolated specific cell populations from Arabidopsis thaliana roots using FACS, and generated tissue-specific transcriptional profiles. These profiles are a valuable resource not only to identify genes with relevant developmental roles, but also for genome annotation allowing the discovery of splicing variants and functional elements.
For MaizeCODE we have extended our techniques to isolate specific cell types in Maize. However, due to the limited number of fluorescent marker lines available, it was not possible to isolate all tissues based on the expression of fluorescent proteins alone. To solve this issue, we developed a combinatorial fluorescent dye technique to differentially label tissue layers in the Maize root. Once labeled, roots were digested and protoplasts separated according to their fluorescent ratio. To calibrate the collection of labeled cells, we made use of an endodermal fluorescent marker line developed by the Jackson lab. This line enabled us to correlate the cell’s dye ratio signal with known positional information.
Isolated cell layers will be used in a set of profiling experiments like RNA-seq, ATAC-seq and ChiP-seq, that will provide enough information to generate a comprehensive catalog of tissue-specific functional elements. In collaboration with the Jackson and Gingeras lab, we have so far generated 6 tissue-specific transcriptional profiles. In addition, we are currently adapting Drop-seq (a high-throughput single-cell seq technique) to be compatible with plant protoplasts. This data will allow us to generate a single-cell resolution transcriptional catalog of the Maize root.
Key personnel: Kenneth Birnbaum (PI), Carlos Ortiz-Ramirez (Postdoc)
Phone: 212-998-8257 | Email: ken.birnbaum@nyu.edu | Website: www.nyu.edu/projects/birnbaum/

Annotation of maize genome function by FACS and ChIP-Seq: To develop functional annotation of the maize genome as a resource for the scientific community, we are profiling B73 cell types by Fluorescence Activated Cell Sorting (FACS) and genome wide transcription Factor (TF) binding by Chromatin Immunoprecipitation-Sequencing (ChIP-Seq). These are important goals of the MaizeCODE project, which is an initial analysis of functional elements in the maize genome.
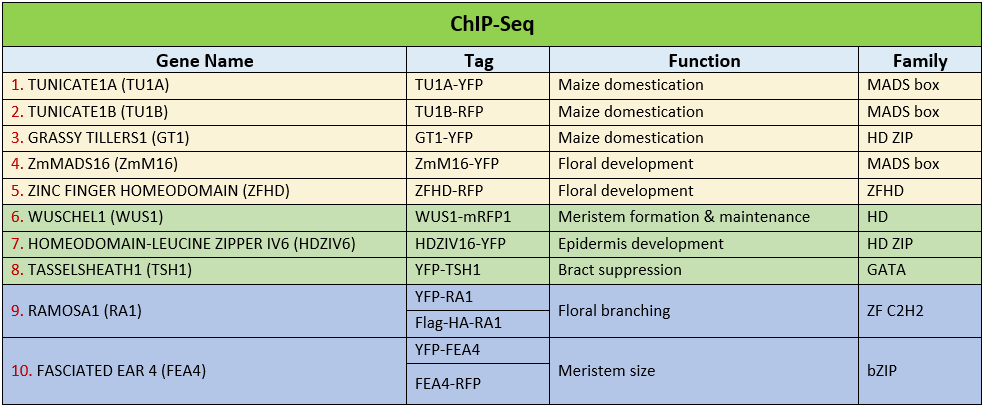
To provide genome wide TF binding site analysis, we are conducting ChIP-Seq for diverse TFs that function in different aspects of maize growth and development. We have generated transgenic lines for these TFs with different tags, such as YFP or RFP. Currently we are working on ~10 TFs that belong to several different families (Table 1). For example, the MADS box TF, TUNICATE1A (TU1A), the ZINC FINGER (ZF) TF, RAMOSA1 (RA1), the bZIP TF, FASCIATED EAR 4 (FEA4), and the GATA TF, TASSELSHEATH1 (TSH1). To overcome limited tissue availability, we are crossing these lines into a branched silkless; Tunicate (bd;Tu) mutant to generate enough inflorescence meristem tissue. Tissues of two of these TFs lines, ZINC FINGER HOMEODOMAIN (ZFHD)-RFP and ZmMADS16 (ZmM16)-YFP have been collected for ChIP-Seq analysis. We are currently working on ChIP using GFP or RFP antibodies to pull down the TF-DNA complex. Future work will use Next Generation sequencing and peak calling to identify the specific binding sites of these TFs.
FACS has provided valuable information, such as cell-type specific mRNA profiling, for the Arabidopsis and rice communities. In maize, we are using fluorescent protein (FP) promoter lines developed by our lab to isolate specific cell types or domains for a subset of profiling experiments, including nuclease sensitivity, PolII, and histone modification. The current FP promoter lines covers many different types of maize cells or tissues (Table 1), such as meristem central zone (pZmWUSCHEL1-mRFP), boundary zone (pZmRAMOSA3-RFP), and root endodermal zone (pZmSCARECROW4-TagRFP). To generate enough tissue for protoplasting and sorting, we are also crossing these FP lines into the bd;Tu mutant. FACS sorting is performed in collaboration with the Birnbaum group. Once cells are harvested and sorted, we will extract the sorted protoplasts for RNA-Seq and chromatin modification and small RNAs analysis.
Key personnel: David Jackson (PI), Xiaosa (Jack) Xu (ChIP-Seq and FACSg)
Phone: 516-367-8467 | Email: jacksond@cshl.edu | Website: jacksonlab.labsites.cshl.edu

Epigenetic mechanisms of gene regulation—chemical and conformational changes to DNA and the chromatin that bundles it—have had an important impact on genome organization and inheritance and on cell fate. These mechanisms are conserved in eukaryotes and provide an additional layer of information superimposed on the genetic code.
Recent improvement in epigenomic profiling technologies have identified a large number of enhancers and verified that they are the most dynamic type of functional elements in the non-coding portion of the genome. Our group is looking into three of the most relevant histone marks associated with active promoters and enhancers: H3K27Ac, H3K4me1, H3K4me3, together with the RNA pol II occupancy. We are working on five different tissues (5-10 mm ears, pollen, 5 days after germination (DAG) coleoptilar nodes, 5 DAG root tips (1-2 mm) and 15 days after pollination (DAP) endosperm) from three maize inbred lines: NC350, W22 and B73, and one inbred teosinte T11. By comparing experimentally defined transcription units with chromatin modifications, we will be able to define combinations that specify functional elements in the maize genome.
Key personnel: Rob Martienssen (PI), Michael Regulski (Chip-Seq)
Phone: 516-367-8322 | Email: martiens@cshl.edu | Website: martienssenlab.labsites.cshl.edu/

Dick McCombie's team has been involved in international efforts culminating in genome sequences for maize, rice, and bread wheat—three of the world's most important food crops. They have also had an important role in projects to sequence the flowering plant Arabidopsis thaliana (the first plant genome sequence), the fission yeast Schizosaccharomyces pombe, as well as the human genome and other important genomes.
McCombie's group is involved in the introduction and optimization of novel methods of high-throughput genome sequencing. For MaizeCODE, we will use the Blue Pippen and our most optimized library prep procedures to make libraries for the target genomes. Following library prep we will load PacBio SMTR cells, using procedure we are currently optimizing. We expect that we will need 60X coverage for each genome to obtain the very long reads (greater than 10kb) that are needed for an optimal assembly.
Key personnel: Richard McCombie (PI), Sarah Goodwin (sequencing)
Phone: 516-422-4083 | Email: mccombie@cshl.edu | Website: mccombielab.labsites.cshl.edu

The MaizeCODE project offers a unique opportunity for students to participate in a big science project. The real-time release of over 150 sequence datasets in 2017-18 will allow students to do de novo explorations of the structure and function of the maize genome. The outreach program will train faculty from primarily undergraduate institutions (PUIs) to use MaizeCODE data and workflows to engage large numbers of students in authentic research—in the context of undergraduate courses. Students will have access to virtual machines in the CyVerse cloud configured to run specific analyses, including measuring differential expression and splicing, identifying promoters and enhancers, and correlating transcription marks with GWAS hits. Of special interest, 60% of participants surveyed at the 2017 Maize Genetics Meeting (n = 112) said they would participate in annotating a gene family of interest or evaluating community annotations; about 40% said they would annotate a gene family for a class project. So CyVerse’s easy-to-use DNA Subway workflow is being enhanced to support the classroom/community annotation of gene families in maize.
Key personnel: David Micklos (PI), Cristina Fernandez-Marco (educator)
Phone: 516-367-5218 | Email: micklos@cshl.edu | Website: www.dnalc.org

Michael Schatz is the Bloomberg Distinguished Associate Professor of Computer Science and Biology at Johns Hopkins University. His research is at the intersection of computer science, biology, and biotechnology, and focuses on development of novel algorithms for comparative genomics, human genetics, and personalized medicine. Professor Schatz’ laboratory will investigate the comparative and functional genomics of the species through their roles in the Data Analysis Center (DAC) and Data Coordination Center (DCC). This will include performing de novo assemblies of the novel genomes and whole genome alignments to characterize structural and SNP differences. They will also assist in the functional genomics analyses, especially RNA-seq analysis to identify and quantify gene expression differences, and several assays (ChIP-seq, Methyl-seq, DNAse-seq) to identify and study regulatory sequences within the maize genomes.
Key personnel: Michael Schatz (PI), Srividya Ramakrishnan (computational analyst)
Phone: 516-367-5218 | Email: mschatz@cshl.edu | Website: schatzlab.cshl.edu

Genome sequences provide the foundation for understanding the developmental program of an organism and how it responds to an environment, it is still a challenge to structurally and functionally annotate a genome reference. This will require the ability to integrate data, between experiments, and across species. Related to the MaizeCode efforts, our focus has been on the development of both genomic and computational resource to support the development and access to reference genomes, and characterization gene networks associated with a plant's development and response to environment. Specifically, we have played a leading role in the first reference assembly and HapMap for maize, recently improved the B73 assembly with PacBio reads, and will lead the computational annotations of the reference assemblies for the MaizeCode project.
Another major objective of the MaizeCode project is to provide the community integrated resources, including valuable data sets, visualization support, and reproducible workflows. To achieve these, we will extend the CyVerse workflow platform, SciApps, to support annotating gene structure and function, expression and epigenetic profiling, and comparative analyses. The SciApps platform will be used to process all MaizeCode datasets. All analysis will be recorded as reproducible workflows and shared with the community. The SciApps platform, powered by CyVerse, allows any community member with a CyVerse account to run MaizeCODE workflows with MaizeCode data or their own data. The final data distribution will be supported through multiple resources, including CyVerse, MaizeGDB, Gramene, NCBI, and SciApps.
Key personnel: Doreen Ware (PI), Liya Wang (platform & computation), Xiaofei Wang (computation), Kapeel Chougule (computation)
Phone: 516-367-6979 | Email: ware@cshl.edu | Website: www.warelab.org


This material is based upon work supported by the
National Science Foundation, Award #1445025